[BC]2 Tutorials and workshops
Tutorials and workshops take place on Monday, September 9 from 9:00 - 16:00 at the University of Basel. The registration fee for tutorials and workshops is 120 CHF, and attendees have to register using the on-line registration system.
Tutorials will provide participants with lectures or practical training covering topics relevant to the bioinformatics field; workshops encourage participants to discuss technical issues, exchange research ideas, and share practical experiences on some focused or emerging topics in bioinformatics.
- T1: Using the Ensembl REST APIs to programmatically access genomic data
- T2: Genomic epidemiology and phylodynamics with Nextstrain
- T3: Introduction to Machine Learning: opportunities for advancing omics data analysis
- T4: Interpretability for deep learning models in computational biology
- T5: Bioinformatics Pipelines for the Analysis of Viral NGS Data
- T7: Analysis of multi-sample multi-condition scRNA-seq datasets
- WS1: EMBO Young Investigator Workshop on Evolutionary and Computational Biology
- WS2: Annotation and curation of computational models in biology
How to get there
Location and Venue
Situated in Switzerland at the heart of Europe, Basel is one of the continent’s most convenient locations for major events. Basel is easily accessible from many European cities by plane or high-speed trains. Approximate travel times to the venue:
Venue
University of Basel
Kollegienhaus
Petersplatz 1
CH-4001 Basel
Train station: Basel Badischer Bahnhof (DB)
Ca. 10 minutes by public transport with bus line 30, exit at "Universität".
Train station: Basel SBBor SBB/SNCF
Ca. 10 minutes by public transport with bus line 30, exit at "Universität".
Basel EuroAirport (BSL)
Ca. 15 minutes by taxi (ca. 40 CHF / 35 EUR ); ca. 20 minutes by public transport (3.80 CHF/ 3.50 EUR). When arriving at Basel airport, take the exit to "Switzerland" (not France). For public transport, take bus line 50 to SBB train station and from there bus line 30 and exit at "Universität".
Zürich Airport (ZRH)
Ca. 90 minutes by train and public transport to the conference site (ca. 45 CHF / 37 EUR). Trains to Basel are leaving from Zürich airport every 30 minutes (either direct or via Zürich main station; train schedules are displayed at the luggage belts. Take the train to either Basel SBB or Basel Badischer Bahnhof.
T1: Using the Ensembl REST APIs to programmatically access genomic data
Room
Seminarraum 106
Organisers and tutors
- Astrid Gall, Ensembl Outreach Team, EMBL-EBI, UK
Overview
The Ensembl project provides a comprehensive and integrated source of annotation of genome sequences, including genes, genetic variation, features that regulate gene expression, homologues and alignments. These data are accessible programmatically in a language agnostic manner via the Ensembl REST APIs. Scripting against public databases like Ensembl facilitates quick retrieval of valuable data in your preferred format or can be integrated into pipelines for data analysis.
This tutorial is aimed at researchers and developers interested in exploring Ensembl beyond the website. The workshop covers how to use the Ensembl REST APIs, including understanding the major endpoints and how to write scripts to call them.
Feedback from previous courses
- “Wish I had taken the course long time ago. Didn’t know before how good and powerful APIs are!”
API workshop, EMBL-EBI, January 2016 - “I really enjoyed the course, and the ENSEMBL API will become a very relevant part of my toolset.”
API workshop, Cambridge, December 2013
Audience and requirements
The tutorial is aimed at bioinformatics and wet-lab researchers who use genomic data and would like to use scripting to access these data directly, including integrating into pipelines.
This is a hands-on training course and participants will need to bring a laptop with WiFi enabled in order to take part. Participants must be able to code in Python, Perl or R. The training will utilise Jupyter Notebooks hosted by Microsoft Azure – to use these all participants will need to have a free Microsoft Account.
Maximum participants: 20
Schedule
| Time | Details |
| 09:00-10:45 | Introduction to Ensembl and its data types |
| 10:45-10:30 | Overview of the Ensembl REST API |
| 10:30-11:00 | Coffee Break |
| 11:00-11:45 | Accessing GET queries with the Ensembl REST API |
| 11:45-12:30 | Decoding json; other content types |
| 12:30-13:30 | Lunch with coffee |
| 13:30-14:30 | Chaining REST queries together |
| 14:30-15:30 | Accessing POST queries with the Ensembl REST API |
| 15:30-16:00 | Rate-limiting |
T2: Genomic epidemiology and phylodynamics with Nextstrain
Room
Seminarrraum 107
Organisers and tutors
- Emma Hodcroft, Biozentrum University of Basel & SIB Swiss Institute of Bioinformatics
- Richard Neher, Biozentrum University of Basel & SIB Swiss Institute of Bioinformatics
Overview
Mutations that accumulate in pathogen genome sequences contain information on the pathogen's evolutionary history and allow us to reconstruct patterns of transmission. Since sequencing capacity and surveillance has increased by orders of magnitude in recent years, data sharing, timely analysis, and dissemination of results has become crucial for harnessing the power of genomics in public health.
Nextstrain is an open-source project for phylodynamic analysis, data integration, and visualization of large data sets of viral and bacterial pathogens. Nextstrain can analyse thousands of sequences within minutes and visualizes the results using an interactive browser-based interface (see Fig. 1). This interface can be used locally on the user's computer or shared on the internet.
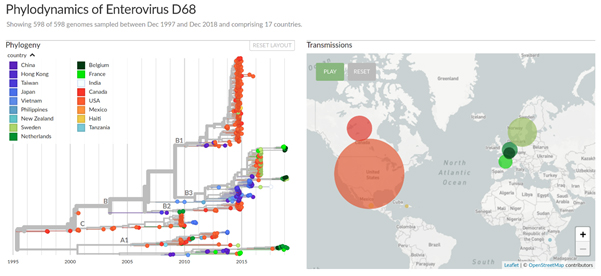
Figure 1: Nextstrain phylodynamic analysis and visualization of Enterovirus D68 in Europe and the US, 2014-2018
Audience and requirements
The workshop is targeted to users with basic bioinformatic knowledge that want to use the Nextstrain pipeline to analyse and visualize their own data. The workshop would be split into a morning session covering background, the structure and use of Nextstrain tools, and tutorials using example data, and an afternoon session where users analyse their own data. Participants would be expected to bring their own laptops. Nextstrain is extensively tested on Linux, MacOS, and Windows 10 with Linux Subsystem and can be installed via conda and npm.
Maximum participants: 23
Schedule
| Time | Details |
| Morning |
|
| Afternoon |
|
T3: Introduction to Machine Learning: opportunities for advancing omics data analysis
Room
Regenzzimmer 111
Organisers and tutors
- Amel Ghouila, Institut Pasteur de Tunis, H3ABioNet
- Fotis Psomopoulos, INAB|CERTH, ELIXIR-GR
Overview
Machine learning has emerged as a discipline that enables computers to assist humans in making sense of large and complex data sets. With the drop-in cost of sequencing technologies, large amounts of omics data are being generated and made accessible to researchers. Analysing these complex high-volume data is not trivial and the use of classical tools cannot explore their full potential. Machine learning can thus be very useful in mining large omics datasets to uncover new insights that can advance the field of medicine and improve health care.
The aim of this tutorial is to introduce participants to the Machine learning (ML) taxonomy and common machine learning algorithms. The tutorial will cover the methods being used to analyse different omics data sets by providing a practical context through the use of basic but widely used R and Python libraries. The tutorial will comprise a number of hands on exercises and challenges, where the participants will acquire a first understanding of the standard ML processes as well as the practical skills in applying them on familiar problems and publicly available real-world data sets.
Learning objectives
- Understand the ML taxonomy and the commonly used machine learning algorithms for analysing “omics” data
- Understand differences between ML algorithms categories and to which kind of problem they can be applied
- Understand different applications of ML in different -omics studies
- Use some basic, widely used Python and R packages for ML
- Interpret and visualize the results obtained from ML analyses on omics datasets
- Apply the ML techniques to analyse their own datasets
Audience and requirements
This introductory tutorial is aimed towards bioinformaticians (graduate students and researchers) familiar with different omics data technologies that are interested in applying machine learning to analyse them.
Prerequisites
- Previous experience in Bioinformatics analysis
- Familiarity with any programming language (especially R) is preferable but not necessary
Maximum participants: 30
Schedule
| Time | Details |
| 09:00 - 09:15 | Tutorial introduction, get to know each other and, Setup |
| Part I: Background | |
| 09:15 - 10:45 | Introduction to ML / DM Data Mining / Machine Learning basic concepts / Taxonomy of ML and examples of algorithms / Deep learning overview |
| 11:00 - 12:30 | Applications of ML in Bioinformatics Examples of different ML/DM techniques that can be applied to different NGS data analysis pipelines / How to choose the right ML technique? |
| Part II: Hands-on | |
| 13:15 - 14:45 | Loading and exploring omics data What is Exploratory Data Analysis (EDA) and why is it useful? Unsupervised Learning How could unsupervised learning be used to analyze omics data? |
| 15:00 - 16:30 | Supervised Learning I: classification How could supervised learning be used to analyze omics data? Supervised Learning II: regression What if the target variable is numerical rather than categorical? |
| 16:30 | Closing, discussion and resource sharing |
T4: Interpretability for deep learning models in computational biology
Room
Hörsaal 117
Organisers and tutors
- María Rodríguez Martínez, IBM Research Zurich
- Matteo Manica, IBM Research Zurich
Helpers in the tutorial
- Ali Oskooei
- Jannis Born
- An-Phi Nguyen
Overview
The recent application of deep neural networks to long-standing problems such as the prediction of functional DNA sequences, the inference of protein-protein interactions or the detection of cancer cells in histopathology images has brought a break-through in performance and prediction power. However, high accuracy often comes at the price of loss of interpretability, i.e. many of these models are built as black-boxes that fail to provide new biological insights. This tutorial focuses on illustrating some of the recent advancements in the field of Interpretable Artificial Intelligence. We will show how explainable, smaller models can achieve similar levels of performance than cumbersome ones, while shedding light on the underlying biological principles driving model decisions. We will demonstrate how to build and extract knowledge using interpretable approaches in two different domains of computational biology, (1) the functional analysis of raw DNA sequencing data and (2) drug sensitivity prediction models. The choice of these two applications is motivated by the availability of adequately large datasets that can support deep learning (DL) approaches and by their high relevance for personalized medicine. We will exploit both publicly available deep learning models as well as in-house developed models.
Learning objectives
The tutorial is aimed to strike the right balance between theoretical input and practical exercises. The tutorial has been designed to provide the participants not only with the theory behind DL and interpretability, but also to offer a set of frameworks, tools and real-life examples that they can implement in their own projects. Specifically, the participants will acquire/refresh basic knowledge on DL models for CB by both a brief technical introduction and a showcase of established models for specific practical applications. Next, several techniques to enhance model interpretability will be explored. In a first case study, a multimodal drug sensitivity prediction model will be introduced and discussed with an emphasis on neural attention mechanism that identify genes and molecular substructures that drove the model decision. Secondly, the problem of predicting transcription factor binding sites from raw DNA sequences will be utilized to demonstrate applications of various interpretability techniques, followed by an evaluation and comparison
Audience and requirements
This course is designed for everyone who would like to learn the basics of interpretability techniques for deep learning. The tutorial will provide a brief introduction to key concepts in deep learning, before exploring recent developments in the field of interpretability. Participants who want to participate in the hands-on exercises should bring a laptop and should have a basic programming knowledge of Python and shell scripting. All the material for the lectures and hands-on exercises will be available prior the day of the tutorial for download.
Maximum participants: 83
Schedule
| Time | Details |
| 9:00 - 9:45 | Introduction to deep learning Deep learning: What, why, how deep? Activation and cost functions, Backpropagation, Regularization, Optimization. |
| 9:45 – 10:30 | Common deep learning models Multi-Layer Perceptron (MLP); Auto-enconders (AE); Convolutional Neural Networks (CNN); Recurrent Neural Networks (RNN). |
| 10:30 – 10:45 | Coffee break |
| 10:45 – 12:00 | Interpretability in deep learning Local versus global methods; A few techniques for interpretability: (1) Perturbation-based approaches, (2) Attention mechanisms, (3) LIME, (4) Surrogate models. |
| 12:00 – 13:00 | Lunch break |
| 13:00 – 14:15 | Drug sensitivity prediction through deep learning and data integration (Hands-on exercise) Introduction to deep learning models for drug prediction; Attention mechanisms to identify genes and structural components relevant for the classification. |
| 14:15 – 14:30 | Coffee break |
| 14:30 – 16:00 | Interpretability for genomic deep learning models (Hands-on exercise) Prediction of transcription factor binding sites using CNNs, Generation of explanations behind the predictions, Evaluation of different methods for interpretability. |
T5: Bioinformatics Pipelines for the Analysis of Viral NGS Data
Room
Seminarrraum 104
Organisers and tutors
- Niko Beerenwinkel, Swiss Federal Institute of Technology Zurich (ETHZ) & SIB Swiss Institute of Bioinformatics
- Susana Posada Céspedes, ETHZ
- Ivan Topolsky, Swiss Federal Institute of Technology Zurich (ETHZ) & SIB Swiss Institute of Bioinformatics
Overview
Viruses are both important models for evolutionary biology and causes of severe infectious diseases, thus representing major public health and economic concern. Viral genetics based on next-generation sequencing (NGS) of viral genomes is now the method of choice for analysing the diversity of intra- and inter-host virus populations, including epidemiological studies and individual treatment optimization in clinical virology.
To support the computational analysis of viral NGS data, we have developed V-pipe, a bioinformatics pipeline that integrates various computational tools. It is freely available at https://cbg-ethz.github.io/V-pipe. It enables the reproducible analysis of genomic diversity in intra-host virus populations, including quality control, read alignment, and inference of viral genomic diversity on the level of single-nucleotide variants and viral haplotypes.
V-pipe uses the workflow management system Snakemake to determine the order in which the steps of the specified pipeline are executed and checks that the output files are produced. It has a modular and extensible architecture. Users can design their own fully reproducible and transparent pipelines. Developers can test their own tools in a defined environment and help establishing best practices for virus research and clinical diagnostics.
This tutorial provides the conceptual basis for analysing viral NGS data and hands-on training on mastering and extending workflows implemented in V-pipe. The purpose of this tutorial is to enable participants to leverage V-pipe to analyse virus sequencing data in production settings, including genomics research and clinical diagnostics.
Learning objectives
- Understanding the general computational concepts behind the most important processing steps and selected software tools implementing them
- Using V-pipe as a practical implementation of those concepts
- Performing quality control of input sequencing data
- Setting up a robust workflow to map reads, call variants, and reconstruct haplotypes
- Use of V-pipe as a benchmarking platform
- Extending and customizing V-pipe
Audience and requirements
The tutorial is accessible to anyone with a Bachelor degree in life sciences, bioinformatics or equivalent, including biomedical scientists with additional (self-)training in bioinformatics. The main target audience of the tutorial are scientists who analyse virus sequencing data either in basic research or clinical settings, e.g., bioinformaticians embedded in clinical research groups, staff of bioinformatics core units, etc. As V-pipe is an open platform, which includes benchmarking capabilities, this tutorial is also of interest to developers who want to test their own tools or integrate those into the pipeline. Both the general concepts and V-pipe are of more general interest to researchers and practitioners, who wish to analyse the genomic diversity of a sample in a reproducible and streamlined manner.
Prerequisites
- Basic UNIX command line, such as provided in SIB's UNIX fundamentals e-learning module.
- Basic knowledge of common sequencing data file formats.
- To simplify organization, we will provide virtual machine images ahead of the tutorial. All participants should bring a laptop and make sure they can run the virtual machine on it.
- A laptop with at least 10 GB free on hard disk and 4 GB of RAM.
Maximum participants: 20
Schedule
| Time | Details |
| Morning | Session 1 - Methodological background I (talks)
|
| Coffee break | |
| Morning | Session 2 - Methodological background II (talks)
|
| Lunch | |
| Afternoon | Session 3 - Basic workflows (hands-on)
|
| Coffee break | |
| Afternoon | Session 4 - Advanced workflows (hands-on)
|
T7: Analysis of multi-sample multi-condition scRNA-seq datasets
Room
Seminarrraum 119
Organisers and tutors
- Mark D. Robinson, Associate Professor, Statistical Genomics, UZH & SIB Swiss Institute of Bioinformatics
- Helena L. Crowell, PhD student, Robinson group, UZH & SIB Swiss Institute of Bioinformatics
- Pierre-Luc Germain, Statistical Genomics, UZH; SIB Swiss Institute of Bioinformatics & Laboratory of Systems Neuroscience, ETHZ
Overview
Single-cell RNA-sequencing (scRNA-seq) has quickly become an empowering technology to characterize the transcriptomes of individual cells. Most early analyses of differential expression (DE) in scRNA-seq data have aimed at identifying differences between cell types, and thus are focused on finding markers for cell sub-populations (experimental units are cells).
There is now an emergence of multi-sample multi-condition scRNA-seq datasets where the goal is to make sample-level inferences (experimental units are samples), with 100s to 1000s of cells measured per replicate. To tackle such complex experimental designs, so-called differential state (DS) analysis follows cell types across a set of samples (e.g., individuals) and experimental conditions (e.g. treatments), in order to identify cell-type specific responses, i.e., changes in cell state. DS analysis: i) should be able to detect “diluted” changes that only affect a single cell type or a subset of cell types; and, ii) is orthogonal to clustering or cell type assignment. Furthermore, cell-type level DE analysis is arguably more interpretable and biologically meaningful.
While there is an opportunity here to leverage existing robust bulk RNA-seq frameworks, by first aggregating single cells into “pseudo-bulk” data at the sub-population level, many new questions arise: How does one track subpopulations across patients (e.g. in the presence of batch effects)? Do we lose information by aggregating (i.e. is it better to model the single-cell data directly)? How do we do normalization when using pseudo-bulks? An extensive workflow for DS analysis in CyTOF data has been established, along with a set of visualisations and differential testing methods, and has been applied to predict cell-type specific responses to immunotherapy. An analogous approach has been applied in the context of scRNA-seq to uncover cell-type specific responses of lupus patients to INF-beta stimulation. However, while there have been comprehensive comparisons of DE analysis methods for scRNA-seq data, an analysis framework for sample-level DE analysis of scRNA-seq data has not been established yet.
Learning objectives
In this tutorial, we will introduce participants to R-based scRNA-seq analysis workflows that are tailored to complex experimental designs. Upon completion, attendees should have become familiar with:
- Basic pre-processing steps (e.g., filtering and normalisation)
- Exploring and evaluating different clusterings
- Identifying cluster-specific marker genes to aid in assigning cell populations
- Aggregation of single-cell to pseudo-bulk data
- DS analysis to detect cell-type specific changes across conditions
- DA analysis to detect differences in cell-type frequencies across conditions
- How to visualise and explore differential testing results
Audience and requirements
Intermediate knowledge of R. Personal laptop with WiFi connection, and latest versions of R, Rstudio and various Bioconductor packages pre-installed.
Maximum participants: 30
Schedule
| Time | Details |
| 09:00 – 10:30 | Introduction, motivation, and historical/theoretical background |
| 10:30 – 11:00 | Coffee break |
| 11:00 – 12:30 | Session 1: pre-processing & clustering |
| 12:30 – 13:30 | Lunch |
| 13:30 – 14:30 | Session 2: DA/S analysis; visualisation and exploration of results |
| 14:30 – 15:30 | Session 3: Do-it-yourself application |
| 15:30 – 16:00 | Closing discussion: Q&A, current state/future directions of the field |
WS1: EMBO Young Investigator Workshop on Evolutionary and Computational Biology
Room
Hörsaal 115
Organisers
- Christophe Dessimoz, University of Lausanne & SIB Swiss Institute of Bioinformatics, Switzerland
- Manuel Irimia, CRG Barcelona, Spain
- Giulia Rancati, A*Star, Singapore
Overview
The goal of this workshop is to foster exchanges on cutting-edge research topics in evolutionary and computational biology. Current and past EMBO Young Investigators & Installation Grantees will give short talks, interspersed by ample time for discussion.
To foster exchanges and interactivity among the participants, attendance to the workshop will be capped at 20 participants (including speakers) - with priority given to early-career researchers aspiring to progress toward independence.
The workshop will include a roundtable discussion on raising the profile of evolutionary and computational biology within the EMBO Young Investigator Programme, as these areas are currently underrepresented in the programme. Potential prospective applicants to the programme are particularly encouraged to join the workshop.
Speakers
- Claudia Bank, Gulbenkian Institute, Portugal
- Christophe Dessimoz, University of Lausanne, Switzerland
- Santiago F. Elena, Spanish National Research Council, Valencia, Spain
- Manuel Irimia, CRG Barcelona, Spain
- Anna Karnkowska, University of Warsaw, Poland
- Tomas Marques Bonet, Universitat Pompeu Fabra, Barcelona, Spain
- Giulia Rancati, A*Star, Singapore
- Christian Schlötterer, Vetmeduni Vienna, Austria
- Joanna Sułkowska, University of Warsaw, Poland
- Dario Valenzano, Max Planck Institute for Biology of Ageing, Cologne, Germany
Maximum participants: approx. 20.
Schedule
| Introduction | |
| 9.00 |
Joanna Sułkowska University of Warsaw Poland |
| 9.30 |
Christian Schlötterer Vetmeduni Vienna Austria |
| 10.00 |
Giulia Rancati A*Star Singapore |
| 10.30 | Coffee break |
| 11.00 |
Manuel Irimia CRG Barcelona Spain |
| 11.30 |
Anna Karnkowska University of Warsaw Poland |
| 12.00 |
Santiago F. Elena Spanish National Research Council Valencia, Spain |
| 12.30 | Lunch |
| 13.30 |
Tomas Marques Bonet Universitat Pompeu Fabra Barcelona, Spain |
| 14.00 |
Dario Valenzano Max Planck Institute for Biology of Ageing Cologne, Germany |
| 14.30 |
Claudia Bank Gulbenkian Institute Portugal |
| 15.00 | Short break |
| 15.15 |
Christophe Dessimoz University of Lausanne Switzerland |
| 15.45 | Roundtable discussion |
| 16.30 | End of workshop |
WS2: Annotation and curation of computational models in biology
Room
Hörsaal 116
Organisers
All organisers are members of the CoLoMoTo/SysMod communities, with experience in workshop organisation.
- Anna Niarakis, Univ Evry, University of Paris-Saclay; FR
- Tomas Helikar, University of Nebraska; USA
- Laurence Calzone, Institut Curie/U900, INSERM/Mines ParisTech; FR
Overview
The fast accumulation of biological data calls for more systematic approaches for their integration, analysis and exploitation. The generation of novel, relevant hypotheses from this enormous quantity of data remains challenging. Logical models have long been used to answer a variety of questions regarding the dynamical behaviours of regulatory networks. As the number of published logical models increases, there is a pressing need for proper model annotation, referencing and curation in community-supported and standardised formats.
In this context, organised by members of the Consortium for Logical Models and Tools (CoLoMoTo – http://colomoto.org) and of the Computational Modeling of Biological Systems Community of Special Interest (COSI) of the International Society for Computational Biology (ISCB) (SysMod - https://sysmod.info/), this workshop aims to review and connect different ongoing projects, bringing together people from different communities involved in modelling and annotation of molecular biological entities, interactions, pathways and models.
Invited speakers
Several experts on data annotation, model curation, and community standard development are invited. In addition, a call for abstract submissions will be issued (see details below).
- Martin Kuiper, Norwegian University of Science and Technology, NO,
- Denis Thieffry, IBESN, Paris, FR
- Rahuman S. Malik Sheriff, Project Leader (BioModels), EMBL-EBI, London, UK
- Cristina Casals, UniProtKB/ Swiss-Prot Biocurator, SIB, Geneva, CH
- Paul Thomas – GO, (LEGO/Noctua), USC, USA,
Selected abstracts
Abstract submissions for oral presentations should be sent to the following addresses anna.niaraki@univ-evry.fr, thelikar2@unl.edu, Laurence.Calzone@curie.fr, with the indication: [BC]2_2019 Workshop Submission.
There will be no poster sessions. Abstracts should include title, author affiliation and contact information and a summary of the work that should not exceed 800 words. In an effort to maintain a balanced women to men ratio, female candidates are strongly encouraged to apply.
Important dates
- Submission Opening date: April 20, 2019
- Submission Deadline (abstract): June 28, 2019
- Notification: July 10, 2018
- Workshop Date: September 9, 2019
Schedule
| Time | Details |
| 9h00-9h10 | Welcome and introduction to the workshop |
| 9h10-10h40 | Session 1 – Data/model curation/annotation and available repositories – Chair Denis Thieffry – Anna Niarakis |
| 9h10-9h30 | Martin Kuiper: Towards a curation platform for causal interaction statements. |
| 9h30-9h50 | Marek Ostaszewski: BioKB and MINERVA: a workflow for curation and quick prototyping of annotated knowledge repositories |
| 9h50-10h10 | Sheriff Malik: Curation and annotation of models in BioModels repository promotes reproducibility and reusability |
| 10h10-10h30 | Cristina Casals: SysVasc Prior Knowledge Network: An example of biocuration for Boolean modelling |
| 10h30-11h00 | Coffee/tea break |
| 11h00-12h30 | Session 2 – Community standards development and interoperability/reusability – Chair Ioannis Xenarios – Laurence Calzone |
| 11h00-11h20 | Denis Thieffry: Computational verification of large logical models - application to the prediction of T cell response to checkpoint inhibitors |
| 11h20-11h40 | Tom Freeman: A graphical and computational model of the renal mammalian circadian clock |
| 11h40-12h00 | Paul Thomas: Gene Ontology Causal Activity Modeling |
| 12h00-12h20 | Anna Niarakis + Vidisha Singh: Automated inference of annotated Boolean models from molecular interaction maps using CaSQ |
| 12h20-12h30 | Wrap up of morning sessions |
| 12h30-13h30 | Lunch break with coffee fix |
| 13h30-14h00 | Round table – Discussion - Exchanges |
| 14h00-15h00 | Session 3 – Tools (I) – Chair Julio Saez Rodriguez |
| 14h00-14h20 | Tomas Helikar: Cell Collective |
| 14h20-14h40 | Laurence Calzone + Gaultier Stoll: MaBoSS ecosystem |
| 14h40-15h00 | Vasundra Touré: The Minimum Information about a Molecular Interaction Causal Statement (MI2CAST): a guideline for the management of molecular causal interaction |
| 15h00-16h00 | Session 4 – Tools (II) – Chair Tomas Helikar |
| 15h00-15h20 | Julio Saez Rodriguez CellNOpt |
| 15h20-15h40 | Aurélien Naldi: The CoLoMoTo Interactive Notebook: Accessible and Reproducible Computational Analyses for Qualitative Biological Networks |
| 15h40-16h00 | Eugenia Oshurko: KAMIStudio |
| 16h00-16h15 | Discussion – Concluding remarks |
Scientific committee
An international scientific committee has been assembled to select the presentations from the call for submissions.
- Claudine Chaouiya, IGC, Oeiras PT & I2M, Aix-Marseille University, FR
- Ioannis Xenarios, Ludwig Institute for Cancer Research, CHUV/UNIL, Lausanne, CH
- Denis Thieffry, École Normale Supérieure, Paris, FR
- Julio Saez Rodriguez, Institute for Computational Biomedicine, Heidelberg University, DE
- Dagmar Waltermath, University Medicine Greifswald, Greifswald, DE
- Benjamin Hall, MRC, Cancer Unit, Cambridge, UK
- Laurence Calzone, Institut Curie / U900 INSERM / Mines ParisTech, FR
- Tomas Helikar, Department of Biochemistry; University of Nebraska; USA
- Anna Niarakis, GenHotel, UEVE, Univ Paris-Saclay, FR